Cystická fibróza
Synonyma v širším slova smyslu
Cystická fibróza, plíce
Angličtina: mukoviscidóza, cystická fibróza
Definice cystické fibrózy
Cystická fibróza je dědičné onemocnění. Dědičnost je lékařsky označována jako autozomálně recesivní. Cystická fibróza (cystická fibróza) není zděděna na pohlavních chromozomech X a Y, ale na autozomálním chromozomu 7.
Přečtěte si náš obecný článek o metabolických poruchách: Metabolické poruchy - co to znamená?
Mutace je na takzvaném genu CFTR. Recesivní znamenalo, že pro vypuknutí choroby musely být přítomny dvě vadné kopie genu. Pokud má osoba zdravé a mutované umístění genu na odpovídajícím chromozomu 7, nemoci nenastane.
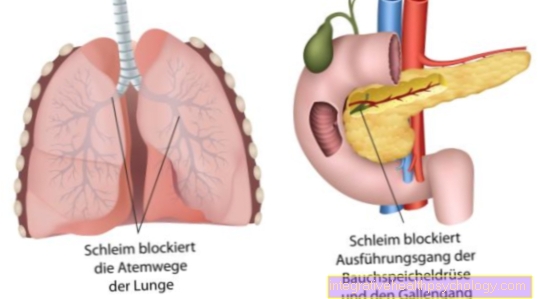
Výsledkem je patologický genový produkt. Tím se kóduje Chloridové kanály jsou rozbité. Vadné chloridové kanály vedou k tvorbě hustého hlenu ve všech exokrinních žlázách.
Tyto exokrinní žlázy, tj. Žlázy, které uvolňují svou sekreci ven, zahrnují:
- slinivka břišní
- tenké střevo
- dýchací systém s plícemi a průduškami
- žlučových cest a
- také Potní žlázy
souhrn
Cystická fibróza je Dědičné onemocnění. Je zděděno takovým způsobem, že je nezávislé na pohlaví a pouze s ním dva vadné geny dojde. Je to nejběžnější autozomálně recesivní dědičnost.
Důsledky jsou tvrdé hlenové útvary všech exokrinních žláz, jako jsou například plíce, pankreas a potní žlázy. Jsou na tom založeni narušený transport chloridu mezi vnitřkem a vně buňky (přečtěte si: Chlorid v krvi). Mutovaný gen je zapnutý Chromozom 7 a způsobuje různé postižení orgánů s odpovídajícími účinky na dýchání, trávení a reprodukci.
Terapie bohužel může zmírnit pouze symptomy, ale nevede k vyléčení. Délka života u pacientů s cystickou fibrózou relativně nízko.
Protože se jedná o recesivní dědičné onemocnění, existují lidé, kteří nesou změněný gen, ale netrpí samotnou nemocí. Takové osoby se nazývají Hlavní dopravce nebo Vodiče, tj. nosiče. Tito lidé nemají cystickou fibrózu, protože druhá kopie genu je neporušená a nemocný není dostatečně silný, aby zvítězil.
Tuto vadnou kopii genu však může předat svému potomstvu. Pokud by již modifikovaný gen stačil k vyvolání nemoci, byla by to takzvaná dominantní dědičnost. Takové dědictví lze nalézt například v Chorea huntington. Více se o této nemoci dozvíte v našem tématu Chorea huntington.
Přibližně 1:2500 leží Míra nemoci u novorozenců v Německu. Dopravce je o všem 25. v německé populaci.
Příčina

Cystická fibróza je způsobena mutací genu na chromozomu 7. Tento chromozom je autozomální chromozom, nikoli pohlavní chromozom.
Každý má 44 autozomálních chromozomů (dvě stejné verze každého) a dva pohlavní chromozomy. Tato mutace na chromozomu 7 vede k tvorbě defektních chloridových kanálů. Reabsorpce (re-absorpce) chloridu z žlázových sekrecí není možná, protože receptor, dokovací místo pro chlorid, není zabudován do žlázových kanálků.
Místo toho je kvůli své nesprávné podobě a struktuře odložena na těžbu. Přirozená výměna chloridu přes určité chloridové kanály je narušena. Tyto takzvané kanály jsou tvořeny proteiny. Na naší DNA je kódováno velké množství proteinů. V důsledku genetické vady chloridových kanálů dochází k dehydratované a tvrdé produkci hlenu ze všech žláz, které uvolňují jejich vylučování ven. Hlen pak částečně blokuje kanály nebo dýchací cesty v plicích.
Přečtěte si o tom také Mutace chromozomu
Diagnóza cystické fibrózy

Typické příznaky začínající v dětství jsou průlomové v diagnostice cystické fibrózy.
Toto podezření je posíleno pozitivní rodinnou anamnézou (nemoc otce / matky nebo blízkých příbuzných). Pozitivní rodinná anamnéza znamená, že v rodině existují nebo již existovaly případy cystické fibrózy - na mateřské nebo otcovské straně.
Ve stolici lze také detekovat nedostatek pankreatických enzymů. Jakékoli ucpání dýchacích cest může být detekováno rentgenováním hrudníku.
Test na pot, který měří obsah chloridů v potu, také pomáhá při diagnostice cystické fibrózy. Pokud je překročena určitá hodnota a platí i další příznaky, diagnóza je relativně pevná. Často si rodiče sami všimnou zvýšeného obsahu soli v potu dítěte.
Nenarozené dítě může být také testováno na toto dědičné onemocnění. Použití vpichu amniotické tekutiny (Amniocentéza) fetální buňky jsou odstraněny a vyšetřeny na mutovaný gen.
Přečtěte si více k tématu: Rentgenové vyšetření dítěte
Terapie cystické fibrózy
Každý, kdo trpí cystickou fibrózou, dostane radu v jednom Cystická fibróza - ambulance nebo radu od Lidský genetik (Specialista na dědičné choroby) doporučeno. Ty mohou pomoci zvýšit kvalitu života nebo, pokud chcete mít děti, vypočítat pravděpodobnost nemocného dítěte. Pokud jsou rodiče plodní a plodní.
Jinak je léčba symptomatická, protože příčinu, vadný gen, nelze vyloučit.
Nevyléčitelná nemoc
Cystická fibróza (cystická fibróza) je dodnes nevyléčitelným onemocněním.
V případě cystické fibrózy je důležité mít dostatečný příjem stolní soli (Chlorid sodný, NaCl). Mukolýza je samozřejmě zaměřena. Mukolýza je rozpouštění hlenu, zejména v plicích, pro usnadnění dýchání.
Léky a inhalace mohou zmírnit příznaky. Pokud se funkce plic znatelně zhoršuje, může být podán kyslík.
Prostřednictvím intenzivní fyzioterapie (fyzioterapie), například masáže klepáním a dechovými cvičeními, se také léčí změny plic způsobené cystickou fibrózou.
Toto onemocnění často končí vyžadovanou transplantací plic. Čekací seznamy jsou však dlouhé.
Součástí terapie je také orální podávání pankreatických enzymů a vitamínů rozpustných v tucích. Úloha slinivky břišní tedy musí být podporována nebo spíše nahrazena. Vitaminy rozpustné v tucích jsou A, D, E a K. Musí být podávány přímo do krve, protože nemohou být absorbovány z potravy kvůli nedostatku trávicích enzymů.
Strava by měla mít také vysoký obsah kalorií, protože pouze zlomek z nich lze získat z jídla.
Aby se předešlo dalším rizikovým faktorům pro komplikace, jako je chřipka nebo pneumonie, mělo by být dítě očkováno. Doporučuje se následující očkování:
- spalničky
- Pneumokoky
- chřipka
Přečtěte si více k tématu: Superinfekce
Tato opatření samozřejmě vyžadují konzultaci s lékařem, s nímž by měla být rizika projednána.
V dnešní době je do genetického výzkumu vložena velká naděje na terapii cystické fibrózy. Pokouší se zavést chybějící genetické informace do lidského genomu. Hledáme vektory, které zvládnou tento úkol. Vektory mohou být například bakteriální nebo virové DNA, které dokážou začlenit zdravou frekvenci do našeho genetického složení.
Terapeutický přístup u nenarozených pacientů je v současné době testován. U myší se myším embryím již podařilo zavést zdravý gen, který obsahoval správnou genovou sekvenci, amniocentézou (inokulace plodové vody). U těchto myší byl tedy produkován zdravý gen CFTR. Amniocentéza je vpíchnutí a odstranění dětských buněk z plodové vody, a to prostřednictvím břišní stěny matky.
V Německu je však tato forma intrauteriny (= v děloze = v děloze) „terapie“ zakázána.
profylaxe
A preventivní opatření v tomto smyslu neexistuje, protože se jedná o dědičné onemocnění.
Lze však navštívit lidské genetické poradenské středisko (obvykle se nachází ve univerzitních nemocnicích). Zde se vypočítává, jak vysoké riziko by bylo přenesení choroby na děti.
Tato rada je vždy užitečná, pokud existuje rodinná anamnéza cystické fibrózy.
Také jeden Prenatální diagnostika stojí za to usilovat. Zde před narozením (tj. Prenatálně) a Vyšetření plodové vody (Amniocentéza) odneseno. Fetální buňky (buňky od dítěte) jsou odebrány z plodové vody a DNA je vyšetřena na mutovaný gen.
Prognóza cystické fibrózy
Průměrná délka života u pacientů s cystickou fibrózou je bohužel pouze 32–37 let. V současné době se odhaduje délka života novorozenců narozených s tímto stavem kolem 45–50 let.
Prognóza do značné míry závisí na terapii a na tom, zda je dodržována.
Samotný pacient a jeho motivace proto hrají důležitou roli.