Chorea huntington
Synonyma v širším slova smyslu
- Vitus tanec (vulg.)
- Huntingtonova nemoc
Angličtina: Huntingtonova nemoc, hlavní chorea.
Definice
Dědičná nemoccož vede ke zničení Mozkové buňky v určitých oblastech mozku nevědomého držení a podpory motorických schopností. Nemoc se obvykle vyskytuje mezi 35-50 lety. Rok života a je vyjádřen v
- Poruchy pohybu jako neúmyslné, bleskově rychlé, smykové pohyby končetin
- Šklebící se
- Degradace intelektuální kapacity a
- Pokles v osobnosti.
Jaká je délka života Huntingtonovy choroby?
Ve srovnání s normální populací je průměrná délka života u pacientů s Huntingtonovou chorobou významně snížena. Jak vysoká je celková délka života, se u jednotlivých osob velmi liší. To záleží na jedné straně na věku nástupu a na druhé straně na průběhu nemoci. První příznaky se obvykle objevují ve věku 30 až 40 let. Téměř polovina postižených zemře během prvních 10 let nemoci. Po 15. roce nemoci je naživu jen 25%. V 10% případů však tato choroba také trvala déle než 20 let. Ženy mají v zásadě o něco delší dobu trvání nemoci než muži. Čím dříve se nemoc objeví, tím vážnější je průběh. Průměrná délka života pacientů s Huntingtonovou nemocí je v průměru mezi 40 a 50 lety, i když s pozdějším nástupem choroby lze dosáhnout věku kolem 60 let.
Epidemiologie:
Frekvence Huntingtonovy choroby je uvedena jako 5 - 10/10 000, dědičnost je autozomálně dominantní. To znamená, že děti postižených mají 50% riziko rozvoje samotné choroby.
Příznaky:
U postižených dochází k uvolnění svalů a současně k expanzi, pohyby podobné blesku končetin, které se zhoršují, když je emoční napětí a zřídka se objevují během spánku.
Důvodem je selhání nezbytných impulsů k potlačení pohybu. Narušená koordinace pohybů se dále projevuje grimasováním, poruchami polykání a obtížemi řeči. Chorea huntington postupuje, jak pacient postupuje, má potíže s chůzí, koordinací pohybů očí a stává se neschopným držet stolici a moč.
Vyskytuje se také u chorea Změny osobnosti jako jsou záchvaty hněvu a poruch pozornosti, jakož i podvody v souvislosti s psychózami. Pokles intelektuálního výkonu vede k progresi demence (Získané mentální postižení, viz tam). Huntingtonova choroba je fatální během 15-20 let od diagnózy, často v důsledku sekundárních onemocnění způsobených špatným celkovým stavem pacienta.
Jaké jsou první známky?
První známky Huntingtonovy choroby jsou obvykle pozorovány ve věku mezi 30 a 40 lety. Psychologické obtíže často předcházejí pohybovým poruchám charakteristickým pro tuto nemoc v letech. Typickými psychologickými abnormalitami jsou deprese a snížená jízda. Počáteční kognitivní deficity se někdy projevují ve formě poruch koncentrace a paměti. Tyto příznaky lze snadno zaměnit za depresi v raných stádiích. Skutečnost, že nemoc často vede k impulzivnímu a škodlivému chování vůči ostatním lidem, je také pro příbuzné stresující.
Pacienti mohou dostávat částečně vizuální informace, např. Výrazy obličeje, již se nezpracovávají správně, a proto již nereagují přiměřeně na emoce ostatních. Poruchy pohybu jsou zpočátku charakterizovány Hyperkinezie (Řecký hyper - asi, kinesis - pohyb). To znamená zvýšené nežádoucí pohyby. Svalový tón - stav napětí ve svalech - je snížen. Pacienti považují tento nedostatek kontroly nad vlastním tělem za velmi stresující. Občas, zejména v rané fázi Pokusy o sebevraždu.
Jak tato nemoc postupuje?
Huntingtonova nemoc je jedna chronicky progresivní neurodegenerativní Nemoc. To znamená, že obvykle postupuje pomalu, ale nepřetržitě, ničí nervy a nakonec vede ke smrti pacienta. Kromě psychologických abnormalit jsou pro nemoc charakteristické také poruchy pohybu. V počátečních stádiích je obvykle více nežádoucích pohybů (Hyperkinezie). Postupem času se člověk vyvíjí Hypokineze. Doslovně přeloženo, to znamená „méně cvičení“, co se míní jako nedostatek pohybu, jak je typické u Parkinsonovy choroby. Jak nemoc postupuje, pacient stále více potřebuje péči. Progresivní demence zpočátku vede k ochuzování jazyka a dezorientaci. Příjem potravy je obvykle ztížen poruchami polykání a pacienti zhubnou. V průměru umírají pacienti 10-15 let po nástupu nemoci. Pokud se nástup onemocnění objeví pozdě, průběh nemoci je často poněkud opožděný.
Existuje lék?
V současné době neexistuje žádný lék na Huntingtonovu chorobu. Od roku 1993 víme, že příčinou onemocnění je vadný gen Chromozom 4. Bohužel v současné době neexistuje žádný způsob, jak zacházet s genetickou vadou nebo jejími důsledky. Proto nemůžete v této chvíli průběh nemoci zastavit. Samozřejmě existuje intenzivní výzkum nových terapeutických přístupů. Genetický základ onemocnění je nyní dobře znám.Z tohoto důvodu mohou postižené osoby a jejich příbuzní jen doufat, že výzkum v určitém okamžiku učiní důležitý průlom.
Které léky pomáhají?
Huntingtonova nemoc je způsobena genovou mutací. Bohužel v současné době neexistují žádné léky, které by tuto příčinu léčily nebo léčily. Jeden může zkusit léčit různé příznaky léky. Neuroleptika se často používají proti klasickým pohybovým poruchám. Antidepresiva pomáhají s depresivními náladami. Nakonec tyto léky nemohou zastavit postup choroby. Snažíte se pomocí léků trochu lépe ovládnout příznaky.
Jak vypadá konečná fáze?
Obvykle je to konečná fáze 10-15 let dosáhl po nástupu nemoci. Pacienti jsou upoutáni na lůžko a potřebují nepřetržitou péči. Protože porucha polykání, která se vyvíjí s postupujícím onemocněním, je mnoho velmi vychudlých (lékařské: kachektický). Při požití jídla také existuje trvalé riziko život ohrožující pneumonie (Aspirační pneumonie) přichází. Pokud pacient již nemůže polykat, je třeba zvážit umělou výživu. Psychologické abnormality se také zvyšují s postupem onemocnění. Nakonec demence prošla, pacienti ztratili schopnost komunikovat a být dezorientováni.
Diferenciální diagnózy
Podobné příznaky, které se skládají z poruch pohybu a intelektuálního úpadku, se mohou objevit v průběhu Creutzfeld-Jakobova nemoc, v pozdějších stádiích nemoci Syfilis a po zánětu Mozek nastat.
Co způsobuje Huntingtonovu nemoc?
Huntingtonova nemoc je genetické onemocnění. Příčinou je genetická vada. Protein (protein), která způsobuje nemoc, se nazývá huntingtin. Gen, který ho kóduje, je na krátkém rameni Chromozom 4. Mutace huntingtinového proteinu způsobuje smrt určitých nervových buněk v určitých oblastech mozku. Jedná se o pomalu postupující proces, a proto je nemoc jednou z takzvaných. neurodegenerativní onemocnění. Mnoho patologických procesů souvisejících s touto chorobou dosud nebylo plně prozkoumáno. Je však známo, že Huntingtonova choroba je jedna Trinukleotidová nemoc jedná. U zdravých lidí se určitá kombinace tří opakuje v DNA až 20krát. U pacientů s Huntingtonovou chorobou se tato kombinace opakuje mnohem častěji, mezi 60 a 250krát. Výsledkem je, že gen již nelze správně přečíst a protein huntingtin je nesprávně sestaven. Čím více se toto opakování vyskytuje, tím dříve se u osoby projeví příznaky. Čím více opakování může být detekováno u pacienta, tím obtížnější je onemocnění.
Diagnóza:
Sbírka lékařské anamnézy a otázek o výskytu Huntingtonovy choroby v rodině. Fyzikální vyšetření se zaměřením na nervový systém.
Měření mozkové aktivity (EEG), případně výpočetní tomografie (rentgenový řez) hlavy. Genetický test, protože základní změny v genetickém materiálu jsou známy, mohou spolehlivě diagnostikovat a dokonce předpovídat Huntingtonovu chorobu. Taková prediktivní (prediktivní) diagnóza je však jen velmi zřídka užitečná, protože nemoc v současné době není léčitelná, a proto by neměly mít žádné terapeutické důsledky.
MRI mozku
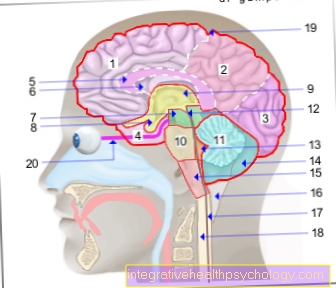
Pokud je podezření na Huntingtonovu chorobu, má smysl nechat si mozek pořídit průřezový obraz. Onemocnění je neurodegenerativní Nemoc, při níž v průběhu procesu umírají nervové buňky v určitých oblastech mozku. To lze také vidět na MRI obrazech. Tkáňová atrofie je zvláště patrná v regionu, který je zodpovědný za dobrovolné hnutí. Takhle Boční komora (= dutiny naplněné mozkovou vodou) rozšířené při zobrazování. Toto je relativně klasický nález Huntingtonovy choroby. Konečná diagnostická jistota je zajištěna genetickým testem (viz oddíl o tomto).
Jak se dědí Huntingtonova nemoc?
Huntingtonova nemoc je jedna autozomálně dominantní zděděná nemoc. Pokud je gen zděděn dominantně, znamená to, že je již vadný Alela na jednom ze dvou Chromozomy vede k charakteristickému výrazu. Termín autozomál je odvozen od autozomů. Všechny chromozomy, které nejsou zapojeny do určování pohlaví, se nazývají autosomy. To znamená, že dědictví je nezávislé na pohlaví. Takže můžete zdědit vadný gen od obou rodičů. Muži i ženy jsou proto postiženi stejně. V případě Huntingtonovy choroby je defektní gen zapnutý Chromozom 4. Ačkoli dědičnost je nezávislá na pohlaví, ukázalo se, že nemoc začíná dříve a má dramatičtější průběh, pokud je vadný gen zděděn od otce. Na druhé straně je v případě dědičnosti matek pravděpodobnější nástup nemoci.
Genetický test
Mutovaný gen, který je zodpovědný za Huntingtonovu nemoc, je vystaven Chromozom 4. Bylo objeveno v roce 1993. Od té doby je k dispozici genetický test. Pokud je pacient podezřelý z Huntingtonovy choroby, může být vyšetřen vzorek krve, aby se zjistilo, zda DNA pacienta má tuto mutaci. To by zajistilo diagnózu. Zdraví lidé, kteří milovali Huntingtonovu chorobu, mohou mít také krevní testy na mutaci. Huntingtonova nemoc je dědičné onemocnění. To má často dalekosáhlé důsledky pro život postižených, a proto existují zvláštní pokyny pro genetické testování u zdravých lidí. Např. nezkoušeli se nezletilí; na žádost třetích stran (rodiče, partneři, ...) nelze provést žádný genetický test. Detekcí genové mutace u zdravých lidí není diagnóza okamžitě diagnostikována, ale pokud je dosaženo určitého počtu opakování určité sekvence v DNA, u postižené osoby se s největší pravděpodobností v průběhu onemocnění objeví Huntingtonova choroba.
Terapie:
Léčba příčiny Huntingtonovy choroby není v současné době možná. Nadměrné poruchy pohybu mohou být potlačeny léky. Za určitých okolností může doprovod psychoterapie nebo připojení ke svépomocné skupině pomoci pacientovi zpracovat znalosti o nemoci.
demence
Kromě klasických pohybových poruch vede Huntingtonova choroba také k psychickým změnám. Jedná se o poruchy Postihnout (= Nálada se mění až do deprese), ale také kognitivní omezení. Ty se často objevují v raných stádiích jako poruchy paměti. Intelektuální schopnosti pacienta jsou na začátku jen nepatrně narušené; to není nutně zaznamenáno zvenčí. Jak nemoc postupuje, dochází k rostoucí ztrátě kognitivních schopností až do demence. Dochází k ochuzování řeči a pacienti jsou často úplně dezorientováni.